Definisi
Dalam bioinformatika, Sequence Alignment (SA) adalah suatu cara untuk mencocokkan sequence DNA, RNA atau protein untuk mengetahui tingkat kemiripan. Jika panjang antara sequences yang dibandingkan tidak sama maka dapat diberikan gaps untuk mengisi kekosongan.

Gambar 1 Sequence Alignment pada Protein
Secara umum, dalam sequences alignment dikenal 2 istilah, yaitu Global Alignment (membandingkan 1 rangkaian sequences secara penuh) dan Local Alignment (hanya mencari subsequences yang mirip).

Gambar 2 Global Alignment

Gambar 3 Local Alignment
Sequences yang sangat pendek atau mirip dapat dicocokkan secara manual, tetapi akan menjadi masalah ketika sequences yang akan dicocokkan sangat panjang. Kemudian berkembanglah beberapa metode untuk sequence alignment.
Metode Sequence Alignment
Untuk membandingkan 2 sequences yang paling dikenal ada 3 metode, yaitu
Metode Dot-Matrixs
Merupakan metode yang paling mudah. Sequences yang dicocokkan dibuatkan sebuah matrix dimana kolom dan baris mereprentasikan masing sequences. Jika ada yang sama (match) maka diberi simbol (dot/titik). Sequences yang berkorelasi akan terlihat membentuk garis di diagonal utama.
Secara visual dapat dilihat fitur dari sequences yang dibandingkan (insertions, deletions, repeats, or inverted repeats).
Masalah dari metode ini adanya noise serta sulit mengekstrak fitur posisi kecocokan antara dua sequences.
S
|
E
|
Q
|
U
|
E
|
N
|
C
|
E
|
A
|
N
|
A
|
L
|
Y
|
S
|
I
|
S
|
P
|
R
|
I
|
M
|
E
|
R
| |
S
|
•
|
•
|
•
| |||||||||||||||||||
E
|
•
|
•
|
•
|
•
| ||||||||||||||||||
Q
|
•
| |||||||||||||||||||||
U
|
•
| |||||||||||||||||||||
E
|
•
|
•
|
•
|
•
| ||||||||||||||||||
N
|
•
|
•
| ||||||||||||||||||||
C
|
•
| |||||||||||||||||||||
E
|
•
|
•
|
•
|
•
| ||||||||||||||||||
P
|
•
| |||||||||||||||||||||
R
|
•
|
•
| ||||||||||||||||||||
I
|
•
|
•
| ||||||||||||||||||||
M
|
•
| |||||||||||||||||||||
E
|
•
|
•
|
•
|
•
| ||||||||||||||||||
R
|
•
|
•
|
Metode Dynamic programming
Metode ini dapat diterapkan untuk mencari Global Alignment maupun Local Alignment dari 2 sequences. Metode yang terkenal adalah Algoritma Needleman-Wunsch (Global) dan Smith-Waterman (Local).
Needleman-Wunsch merupakan metode pertama yang menerapkan Dynamic Programming dalam Sequence Alignment sedangkan Smith-Waterman memodifikasi metode dari Needleman-Wunsch untuk mencari Local Alignment.
Dynamic Programming sangat optimal untuk mencari kecocokan antara 2 sequences dan dapat diperluas untuk lebih dari 2 sequences. Metode ini sangat lambat untuk sequences yang sangat panjang.
Words Method
Dikenal juga sebagai k-tuple methods, merupakan metode heuristic dimana tidak menjamin hasilnya optimal tetapi lebih efisien dibandingkan Dynamic Programming. Sering dipakai untuk pencarian dalam database dengan ukuran yang sangat besar (FASTA, BLAST)
Multiple Sequence Alignment
Dari perbandingan antara 2 sequences, kemudian berkembang menjadi Multiple Sequence Alignment (MSA), yaitu lebih dari 2 sequences yang dibandingkan. Salah satu tujuannya adalah untuk merekonstruksi phylogenetic tree atau untuk mengetahui tingkat kekerabatan spesies.

Gambar 5Phylogenetic Tree
Untuk MSA juga ada 3 metode yang terkenal, yaitu
Dynamic Programming
Secara teori, metode ini dapat mencocokkan banyak sequences tetapi sangat “mahal” dalam segi kompleksitas dan memori. Sama seperti Dynamic Programming yang dijelaskan untuk 2 sequences, algoritma yang terkenal adalah Needleman-Wunsch dan Smith-Waterman.
Progressive Alignment
Cara kerjanya adalah mencocokkan sequence yang terdekat kemudian menambahkan sequence berikutnya sehingga terbentuk tree. Initial tree untuk mencocokkan memakai metode yang mirip dengan FASTA. Contoh metode ini adalah ClustalW dan T-Coffee
Metode ini sangat cepat, namun ketika sudah terbentuk tree tidak bisa menyisipkan sequence yang terbaru yang lebih mendekati kemiripannya. Sehingga keakuratannya tergantung dari inisialisasi awal.
Metode Iterasi
Metode ini mencoba memperbaiki kelemahan dari metode Progressive yang tergantung dari inisialisasi. Metode iterasi mencoba mengoptimalkan berdasarkan fungsi scoring dan dapat mencocokkan ulang (realigning) subset sequence. Metode ini sangat lambat dan hasilnya tergantung probabilitas karena didasari stokastik. Contohya adalah Hidden Markov Models dan Algoritma Genetik.
Online Sequence Alignment
Ada beberapa situs yang menyediakan fasilitas untuk online untuk Sequence Alignment. Contohnya situs yang dikelola European Bioinformatics Institute (http://www.ebi.ac.uk) dan National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). Biasanya jika mempunyai sebuah sequence, dapat melakukan Sequence Alignment pada situs tersebut untuk mencari kekerabatan yang paling dekat yang ada di databasenya.
Universitas Indonesia Culture Collection (UICC) yang dikelola Departemen Biologi FMIPA UI memiliki sistem informasi yang dapat melakukan Sequence Alignment secara online, meskipun dibatasi hanya untuk jaringan intranet terkait dengan hak cipta pada koleksi databasenya. UICC sejak tahun 1980 sampai sekarang telah memiliki sekitar 2.155 strain (sequence) indigenos Indonesia yang terdiri dari 43 strain bakteri; 479 strain kapang (filamentous fungi/moulds) dan 1.933 strain khamir (yeasts).
Sistem informasi yang dikembangkan menggunakan Apache-PHP-MySQL dan untuk melakukan proses Alignment menggunakan metode Needleman-Wunsch yang ada pada bioinformatics toolbox Matlab.
Algoritma Needleman-Wunsch
Saul B. Needleman dan Christian D. Wunsch memperkenalkan algoritma ini pada tahun 1970 dan merupakan Sequence Alignment yang menggunakan Dynamic Programming.
Sama seperti dengan algoritma dynamic lainnya, Needleman-Wunsch membuat tabel (matrix) yang berisi nilai (scoring) kecocokan dari tiap elemen pada sequence.
Langkah pada Needleman-Wunsch untuk proses Alignment antara Sequence X dengan Y adalah sebagai berikut
Initialization
Membuat score matrix F berukuran n x m dimana n, m merupakan panjang dari masing-masing sequences. Untuk inisialisasi matrix F dapat dibuat sebagai berikut
F(0,0)=0
F(0,i)= -i*d
F(j,0)= -j*d
d : cost dari gap pinalty
Scoring
Untuk elemen yang lainnya diisi dengan cara sebagai berikut
F(i,j)=max{F(i – 1, j – 1) + S(xi,yj) F (i – 1, j) – d F (i, j – 1) – d
S(xi,yj)=1 jika xi= yj (match), jika tidak S(xi,yj)=0. Berikut pseudocode untuk mengisi nilai matriks F
for i=0 to length(X)
F(i,0) ← d*i
for j=0 to length(Y)
F(0,j) ← d*j
for i=1 to length(X){
for j = 1 to length(Y)
{
Match ← F(i-1,j-1) + S(Xi, Yj)
Delete ← F(i-1, j) + d
Insert ← F(i, j-1) + d
F(i,j) ← max(Match, Insert, Delete)
}
}
Trace back (Alignment)
Setelah matriks F terbentuk maka dilakukan backtracking move dari bawah kanan (max score) matriks F untuk merekontruksi sequence X dan Y. Dengan cara ini akan didapat bagaimana jalur yang ditempuh sehingga menghasilkan nilai yang maksimum di F(n,m). Berikut pseudocode untuk backtracking move
AlignmentX ← ""
AlignmentY ← ""
i ← length(X)
j ← length(Y)
while (i > 0 and j > 0)
{
Score ← F(i,j)
ScoreDiag ← F(i - 1, j - 1)
ScoreUp ← F(i, j - 1)
ScoreLeft ← F(i - 1, j)
if (Score == ScoreDiag + S(Xi, Yj))
{
AlignmentX ← Xi + AlignmentX
AlignmentY ← Yj + AlignmentY
i ← i - 1
j ← j - 1
}
else if (Score == ScoreLeft + d)
{
AlignmentX ← Xi + AlignmentX
AlignmentY ← "-" + AlignmentY
i ← i - 1
}
otherwise (Score == ScoreUp + d)
{
AlignmentX ← "-" + AlignmentX
AlignmentY ← Yj + AlignmentY
j ← j - 1
}
}
while (i > 0)
{
AlignmentX ← Xi + AlignmentX
AlignmentY ← "-" + AlignmentY
i ← i - 1
}
while (j > 0)
{
AlignmentX ← "-" + AlignmentX
AlignmentY ← Yj + AlignmentY
j ← j - 1
}
Dari pseudocode secara keseluruhan dapat dilihat Algoritma Needleman-Wunsch memiliki kompleksitas kuadratik O(nm).
Contoh Needleman-Wunsch
Akan dilakukan proses Sequence Alignment antara
X = ATCG (n=4, length of sequence X)
Y = TCG (m=3, length of sequence Y)
Initialization
Skema Scoring :
- Match Score = +1
- Mismatch Score = -1
- Gap penalty = -1
Buat matriks dengan n+1 baris dan m+1 kolom. Isi baris dan kolom pertama dengan perkalian gap pinalti.
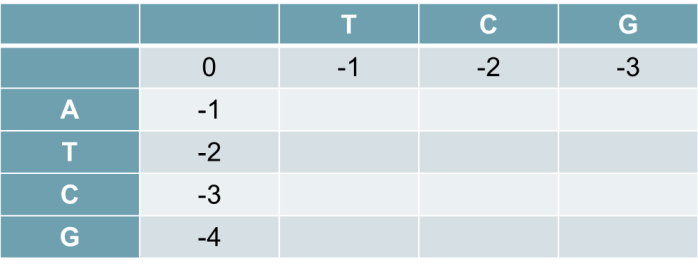
Scoring
Untuk baris-kolom terakhir akan memiliki nilai maksimum, dalam hal ini 2 setelah dijalankan algoritma pengisian matriks.

Traceback
Untuk traceback akan dimulai dari kanan bawah untuk mencari tetangga (top-left, up atau left) yang merupakan predessessornya.
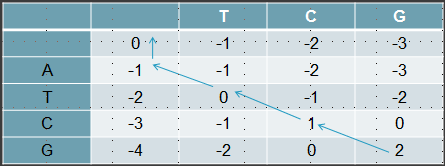
Hasil akhir akan menghasilkan alignment 
A T C G
| | | |
_ T C G
Penutup
Algoritma Needleman-Wunsch merupakan algoritma untuk Global Sequence Alignment. Algoritma ini berdasarkan Dynamic Programming dan memiliki kompleksitas O(nm). Needleman-Wunsch dapat juga dipakai untuk Multiple Sequence Alignment.
Untuk speed up dynamic programming pada Needleman-Wunsch, saat ini dapat digunakan The Four-Russian Algorithm yang memiliki kompleksitas O(N2/log2N).
Sequence Alignment sendiri pernah disinggung sekilas ketika presentasi pertama kelompok yang membahas String Matching.
Daftar Pustaka
Dr G Sudha Sadasivam, & G Baktavatchalam. (2010). A Novel Approach to Multiple Sequence Alignment.
http://en.wikipedia.org/wiki/Needleman-Wunsch_algorithm. (n.d.).
http://en.wikipedia.org/wiki/Sequence_alignment. (n.d.).
Murphy, R. F. (2009). Sequence Database Searching. http://www.cmu.edu/bio/education/courses/03510/LectureNotes/LecturesPart06.ppt.
Pooja Anshul Saxena. (n.d.). The Needleman-Wunsch and The SMITH-WATERMAN Algorithm for Sequence Alignment. http://home.olemiss.edu/~pasaxena/Needleman-Wunsch.ppt.
Serafim Batzoglou. (2003). Sequence Alignment. http://ai.stanford.edu/~serafim/cs262/Spring2003/Slides/Lecture3.ppt.
Thompson, S. M. (2003). An Introduction to Bioinformatics. http://bio.fsu.edu/~stevet/BSC5936/DotMatrix.ppt.
Urmila Kulkarni-Kale. (2010). Pairwise & Multiple sequence alignments. http://sta.uwi.edu/fsa/dms/icgeb/documents/sequence-alignments.ppt.
Wellyzar Sjamsuridzal, Sitaresmi, Gatot F. Hertono, & Ariyanti Oetari. (2006). PENGEMBANGAN DATABASE MIKROORGANISME INDIGENOS INDONESIA. MAKARA Seri Sains Volume 10 No.1 April 2006 ISSN 1693-6671.
Komentar
Posting Komentar